Machine learning in drug design and discovery
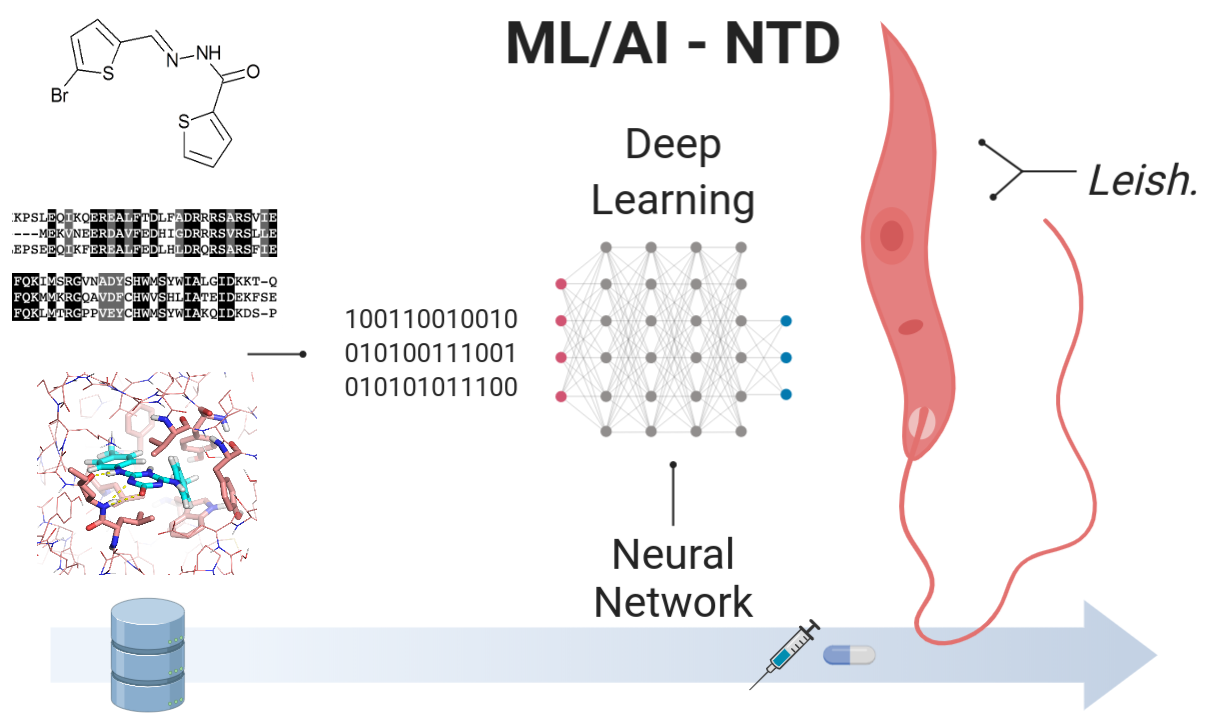
Peña-Guerrero J., Nguewa P.,* García-Sosa A. T.* "Machine Learning, Artificial Intelligence, and Data Science Breaking into Drug Design and Neglected Diseases" WIREs Computational Molecular Science 2021, e1513, Published online 5th January 2021, [HTML], [DOI], [PDF]
García-Sosa A. T.* "Androgen Receptor Binding Category Prediction with Deep Neural Networks and Structure-, Ligand-, and Statistically-Based Features" Molecules 2021, 26:1285 [DOI], [PDF]
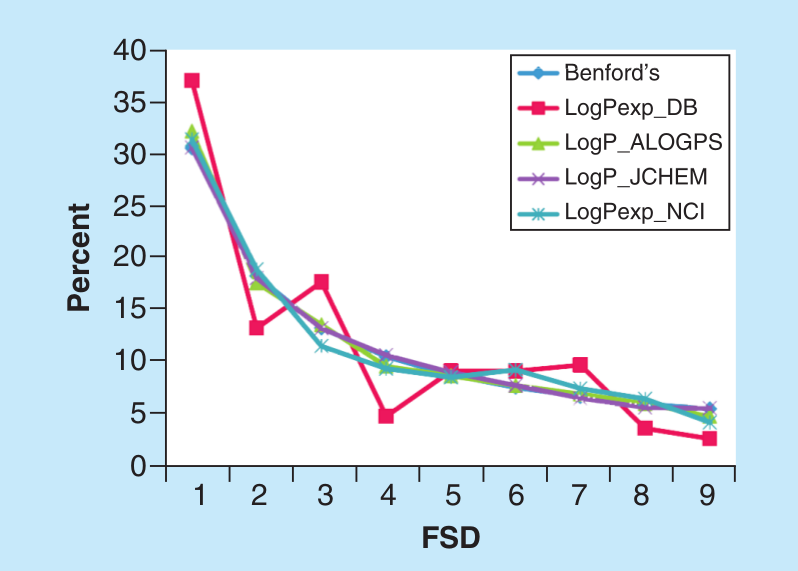
García-Sosa A. T.* "Benford's Law in Medicinal Chemistry: Implications for Drug Design", Future Medicinal Chemistry, 2019, Vol. 11, Iss. 17, 2247-2253, [DOI], [HTML], [PDF], [Data]
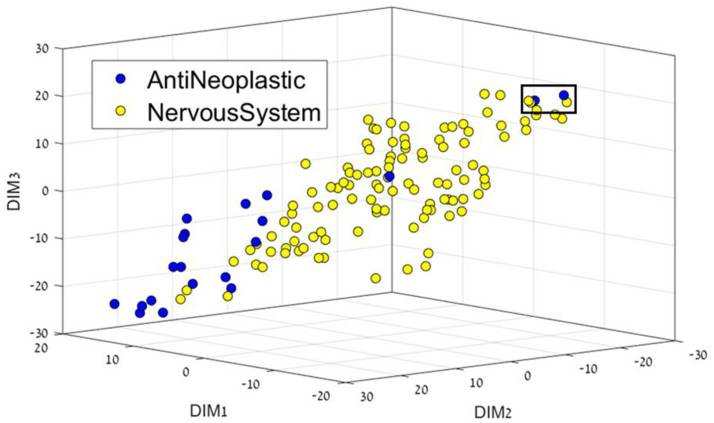
Yosipof A., Guedes R. C., García-Sosa A. T.* "Data Mining and Machine Learning Models for Predicting Drug Likeness and their Disease or Organ Category", Frontiers in Chemistry, 2018, Vol. 6, 162, [DOI], [PDF] Highlighted in the Specialty News. Technology Networks "Data Visualization in Biopharma: Leveraging AI, VR, and MR to Support Drug Discovery"
See Publications.
Inhibitors of Leishmania
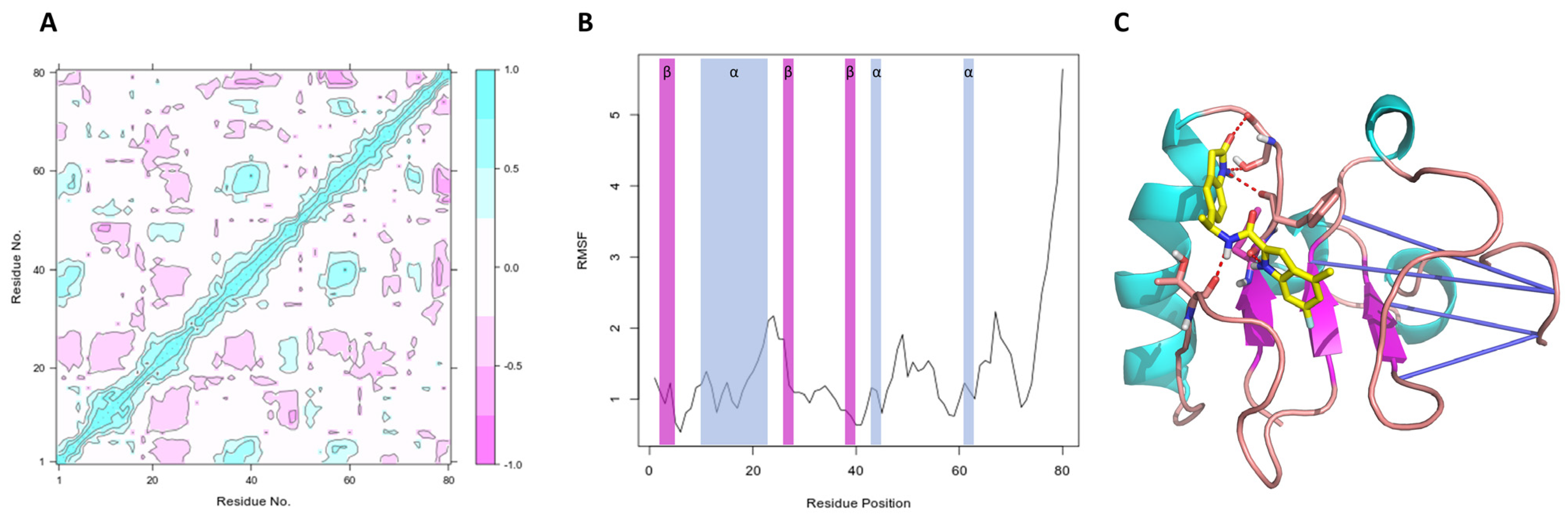
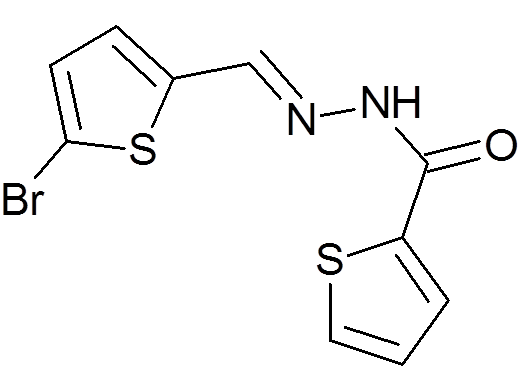
Peña-Guerrero J., Fernández-Rubio C., Burguete-Mikeo A ., El-Dirany R., García-Sosa A. T.*, Nguewa P.* "Discovery and Val idation of Lmj_04_BRCT Domain, a Novel Therapeutic Target: Identification of Can didate Drugs for Leishmaniasis" International Journal of Molecular Sciences 2021, 22:10493 [DOI], [PDF]
Stevanovic S., Sencanski M., Danel M., Menendez C., Belguedj R., Bouraiou A., Nikolic K., Cojean S., Loiseau P. M., Glisic S., Baltas M., García-Sosa, A. T.* "Synthesis, In Silico, and In Vitro Evaluation of Antileishmanial Activity of Oxadiazoles and Indolizine_Containing Compounds Flagged Against Antitargets", Molecules, 2019, Vol. 24, Iss. 7, 1282, [DOI], [HTML], [PDF]
García-Sosa A. T.* "Designing Ligands for Leishmania, Plasmodium, and Aspergillus N-Myristoyl Transferase with Specificity and Antitarget Safe Virtual Libraries", Current Computer-Aided Drug Design, 2018, Vol. 14, No. 2, 131-141, [DOI], [PubMed]
Glisic S., Sencanski M., Perovic V., Stevanovic S., García-Sosa A. T.* "Arginase Flavonoid Antileishmanial In Silico Inhibitors Flagged Against Antitargets" Molecules 2016, 21(5):589 [HTML], [DOI], [PDF]
See Publications.
Cobalt-Ligand complexes inhibit bacterial film and quorum sensing
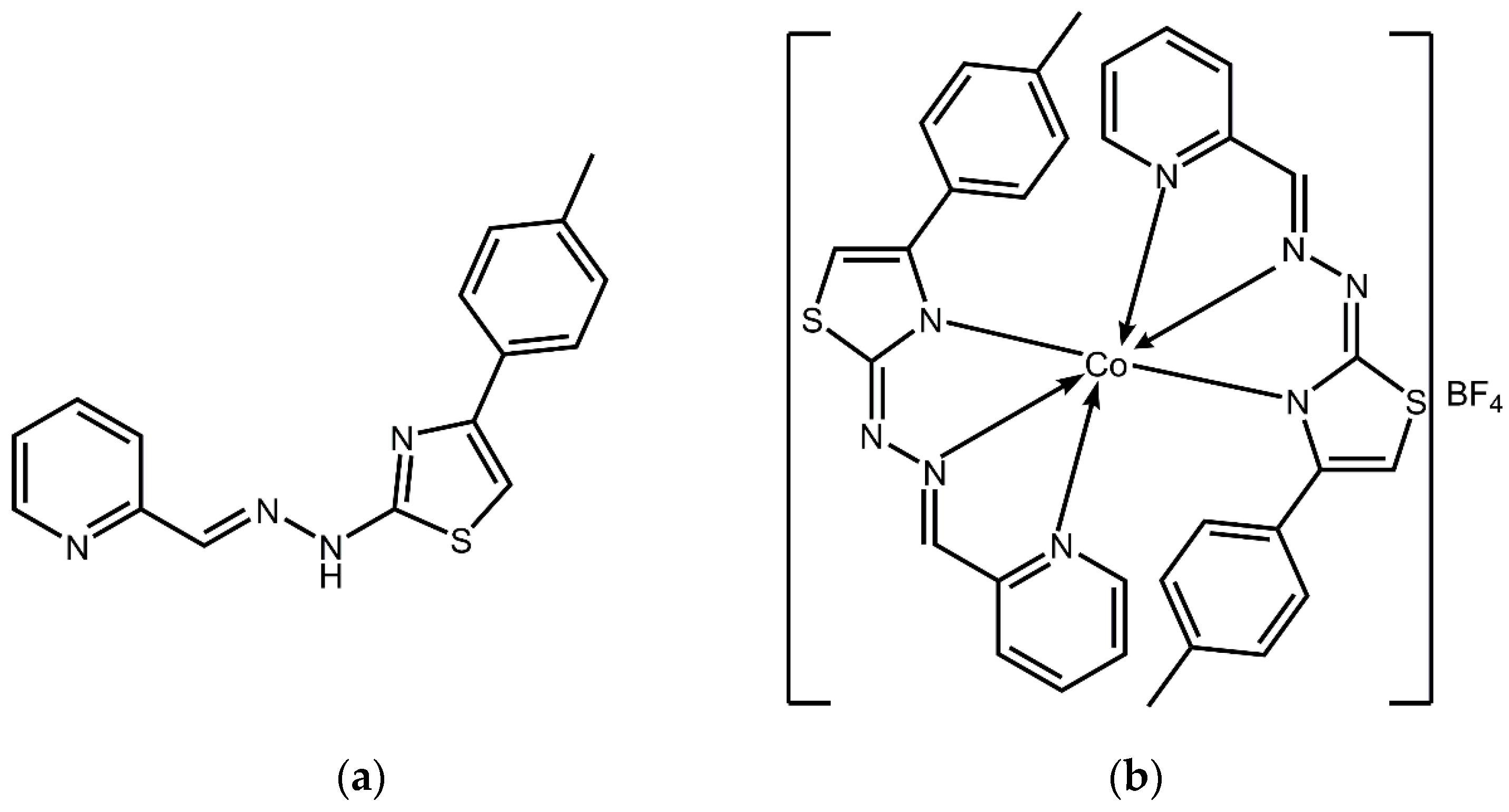
Borges A., Simões M., Todorovic T. R., Filipovic, N. R., García-Sosa A. T.* "Cobalt Complex with Thiazole_Based Ligand as New Pseudomonas aeruginosa Quorum Quencher, Biofilm Inhibitor, and Virulence Attenuator", Molecules, 2018, Vol. 23, 1385, [DOI], [HTML], [PDF]
See Publications.
Delivering DNA in vivo with cell penetrating peptides
Selected for Cover Figure and Editorial Comment

Freimann K., Arukuusk P., Kurrikoff K., Ferreira Vasconcelos L. D., Veiman K. -L., Uusna J., Margus H., García-Sosa A. T., Pooga M., Langel Ü, "Optimization of in vivo DNA delivery with NickFect peptide vectors", Journal of Controlled Release, 2016, Vol. 241, 134-143. [HTML], [DOI]
See Publications.
Cell-penetrating peptides binding to siRNA
The binding affinity of a series of cell-penetrating peptides (CPP) was modeled through docking and making use of the number of intermolecular hydrogen bonds, lipophilic contacts, and the number of sp3 molecular orbital hybridization carbons. The new ranking of the peptides is consistent with the experimentally determined efficiency in the downregulation of luciferase activity, which includes the peptides' ability to bind and deliver the siRNA into the cell. The predicted structures of the complexes of peptides to siRNA were stable throughout 10 ns long, explicit water molecular dynamics simulations. The stability and binding affinity of peptide-siRNA complexes was related to the sidechains and modifications of the CPPs, with the stearyl and quinoline groups improving affinity and stability. The reranking of the peptides docked to siRNA, together with explicit water molecular dynamics simulations, appears to be well suited to describe and predict the interaction of CPPs with siRNA.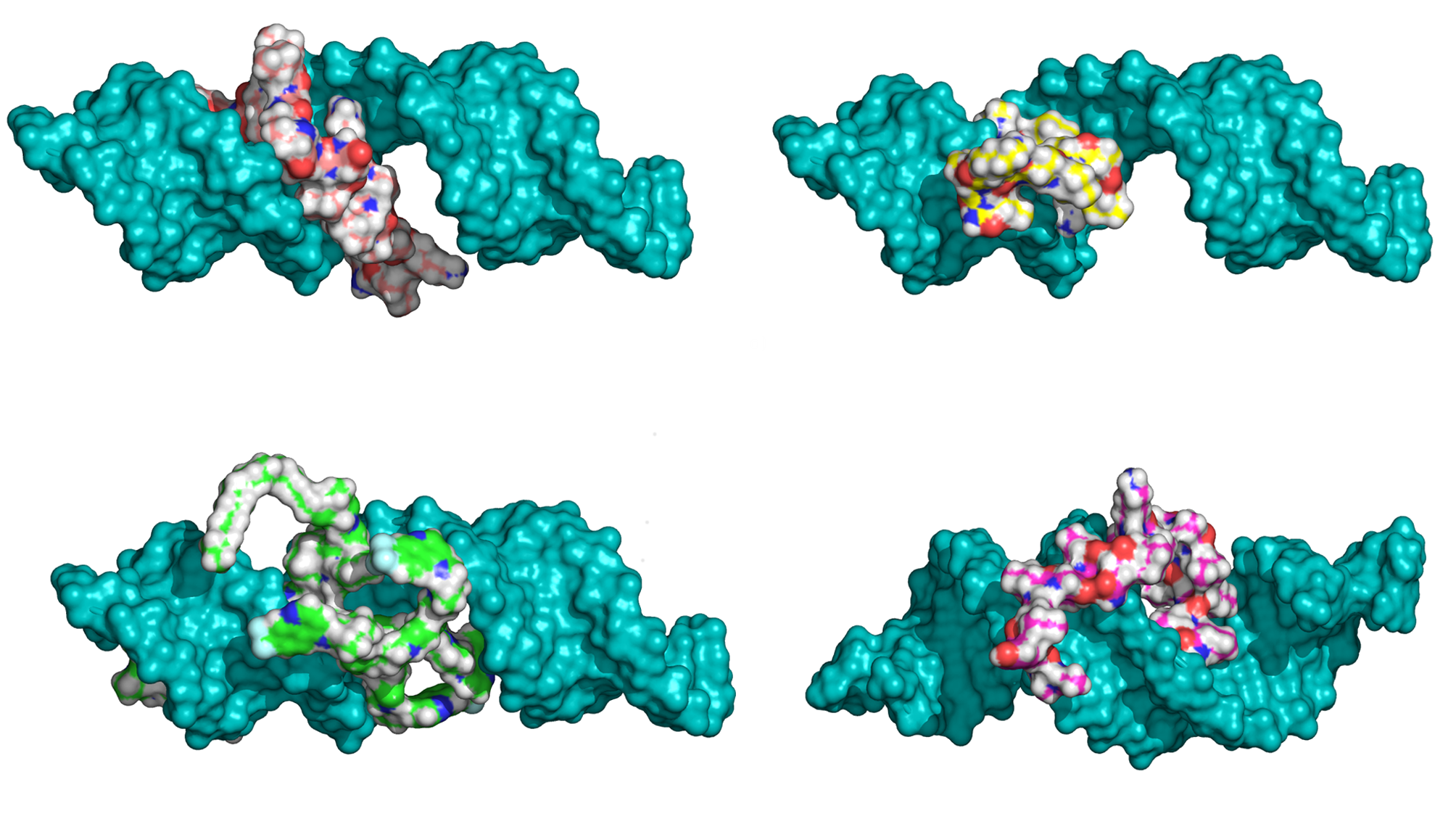
García-Sosa A. T.,* Tulp I., Langel K., Langel Ü., "Peptide-Ligand Binding Modeling of siRNA with Cell-Penetrating Peptides", BioMed Research International, 2014, Vol. 2014, Article ID 257040, 7 pages. DOI: http://dx.doi.org/10.1155/2014/257040, [PDF].
See Publications.
Mutations in MPK12 produce different activity in plant signalling
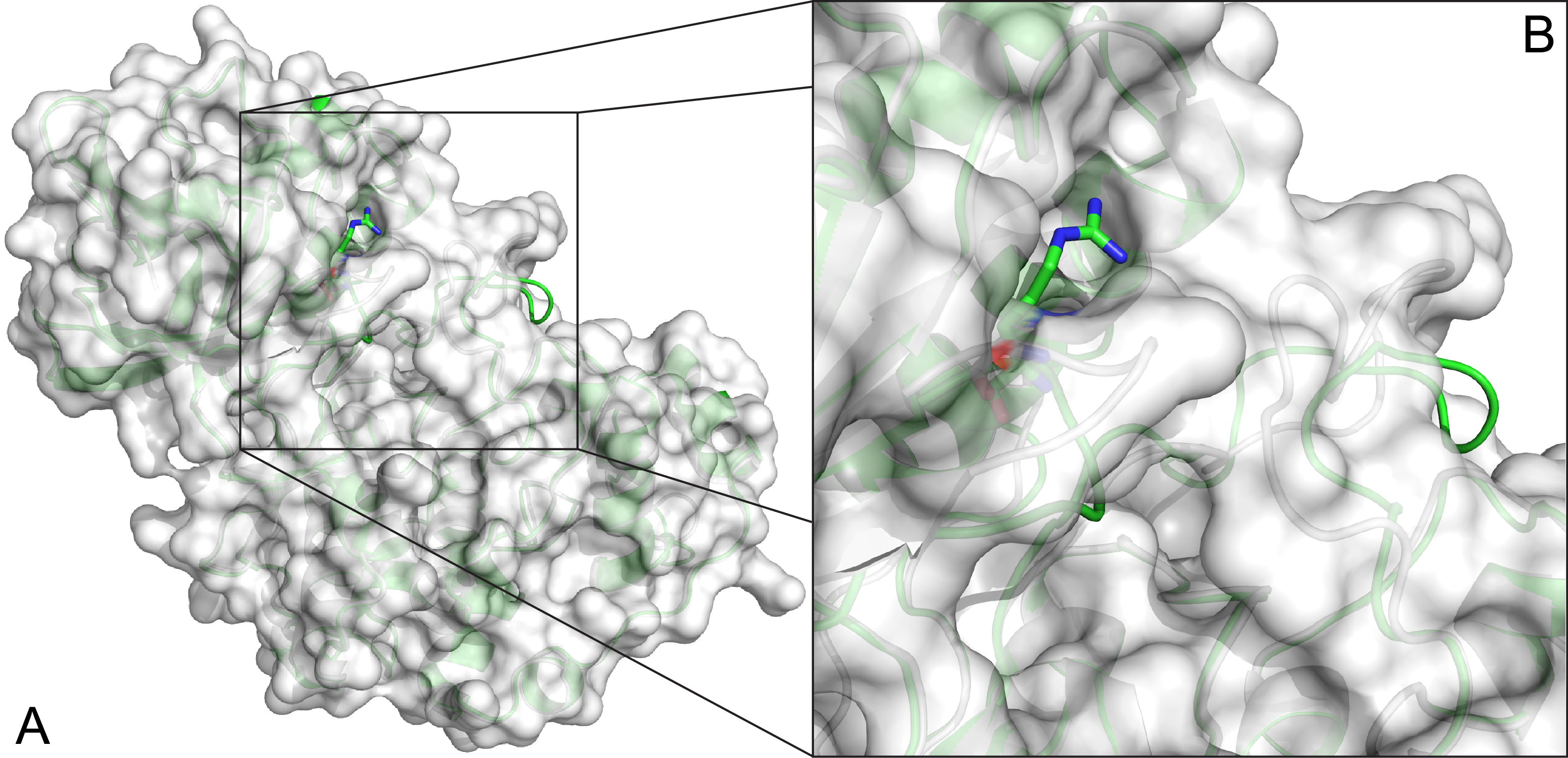
See Publications.
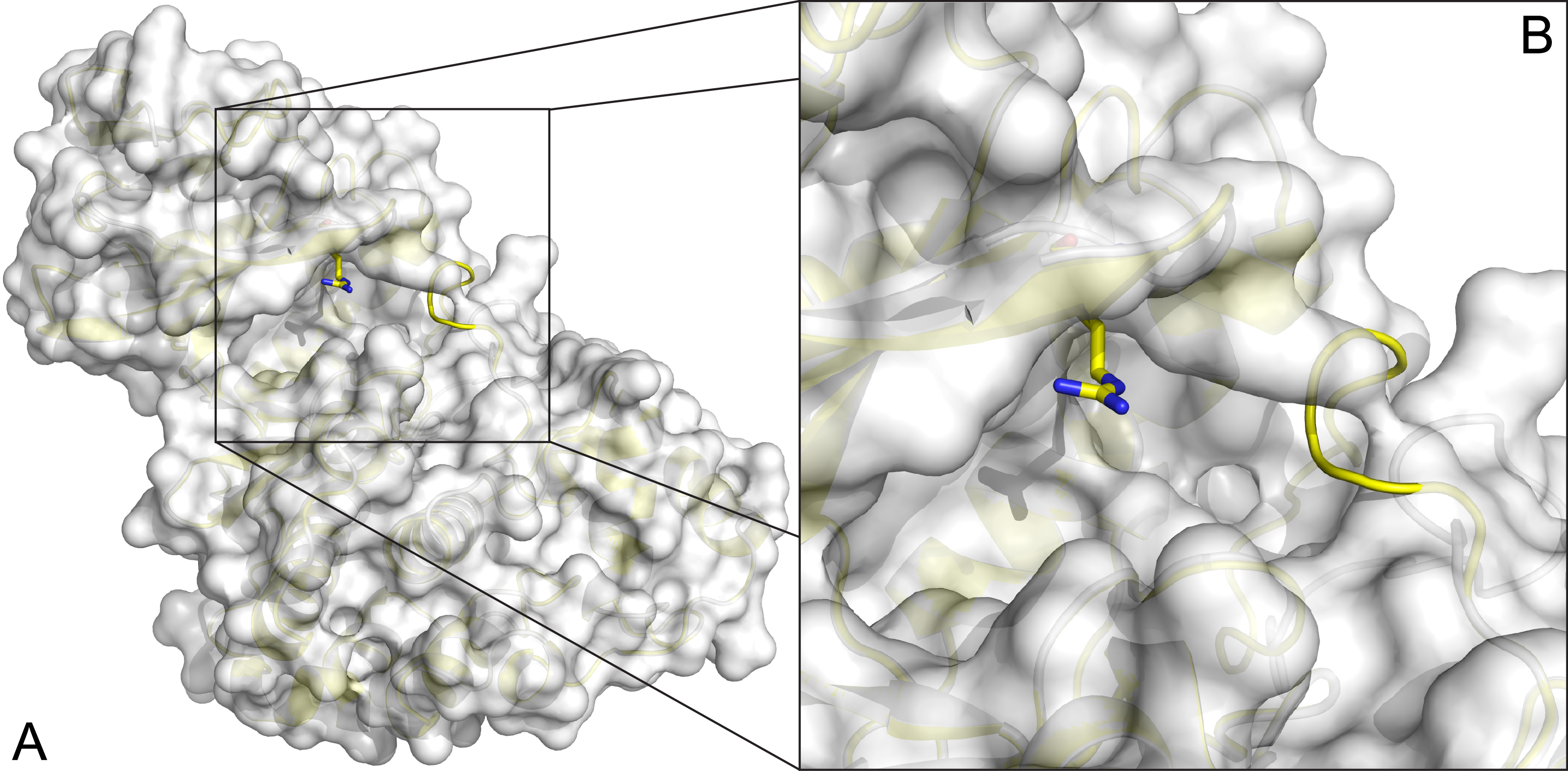
Small, low-toxicity, novel compounds that inhibit HIV-1
See Publications.
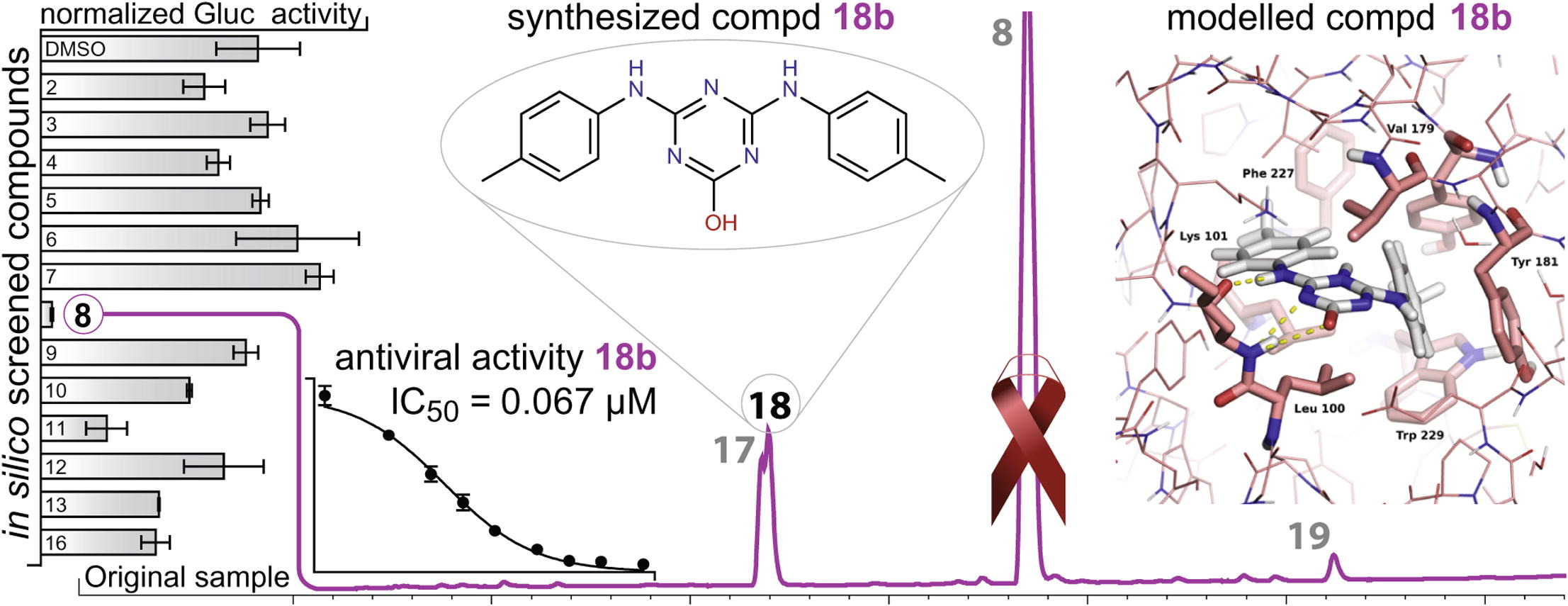
Virtual Screening for HIV-1 Protease
See Publications.
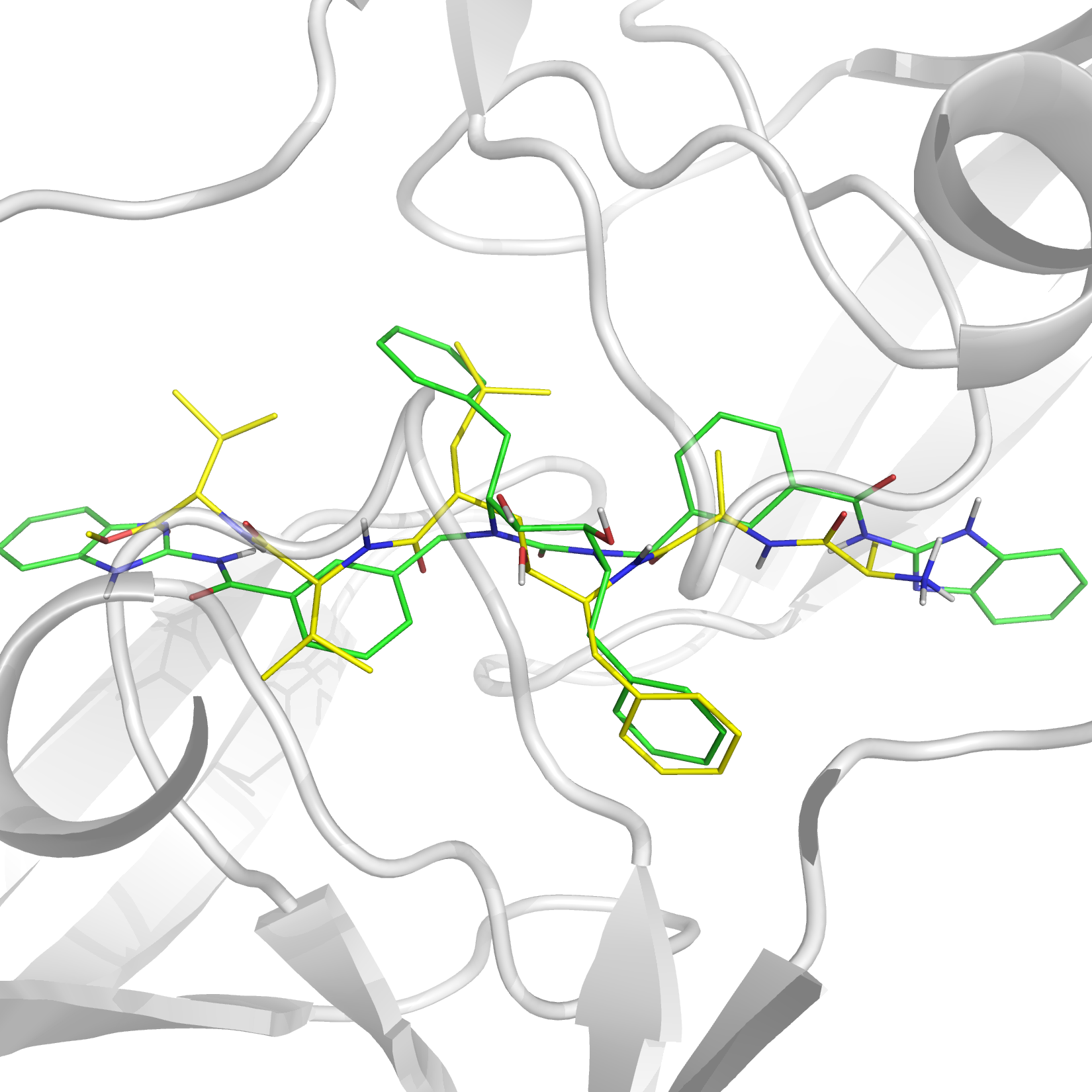
Triangular numbers in re-ranking for HIV-1 Integrase
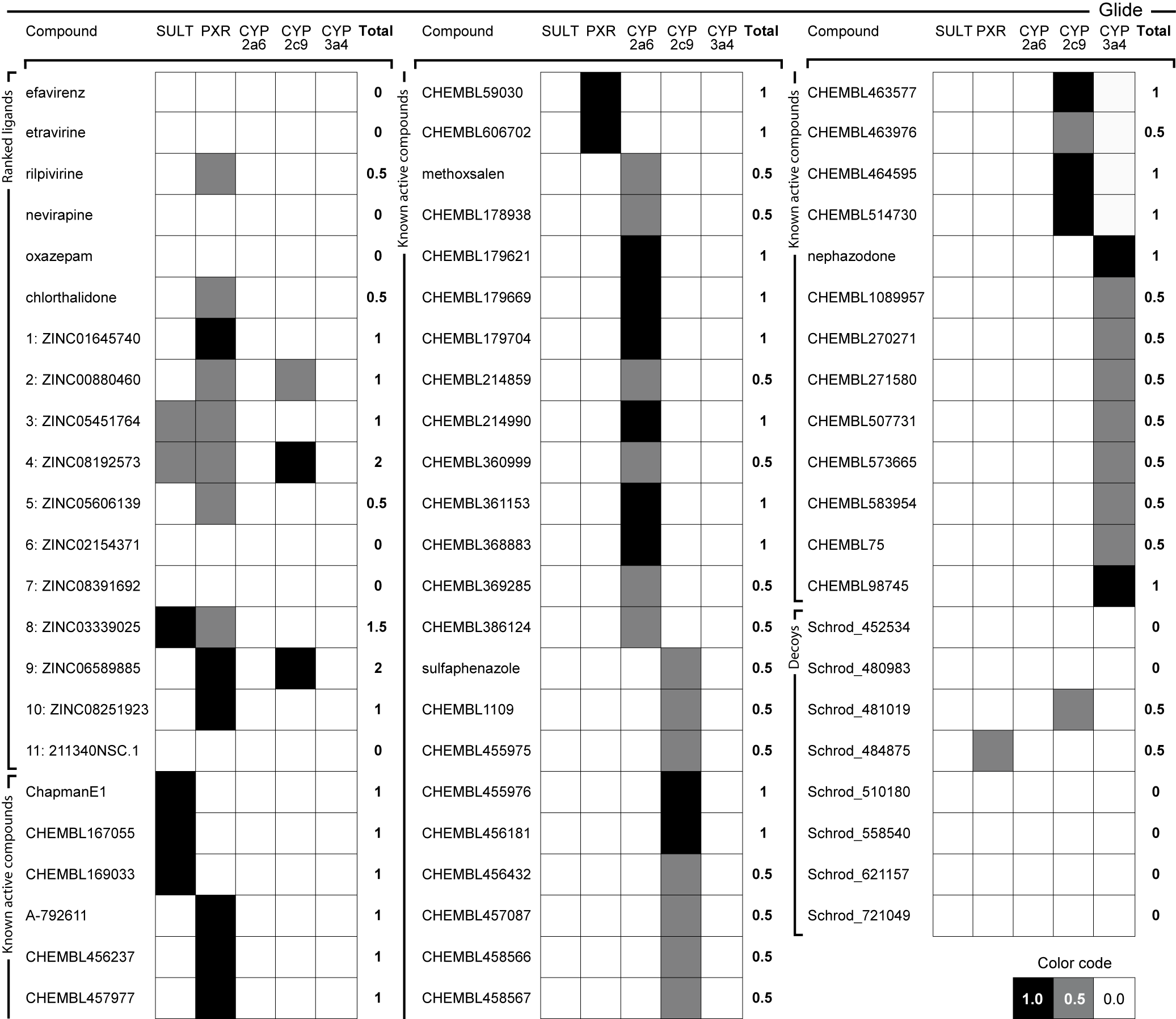
Ranking in virtual screening for HIV-1 integrase was improved using triangular numbers, which can group different runs from mutants, conformations and conditions. They are a growing series of numbers that in this work allow to define an objective threshold above which the majority of the top compounds are known inhibitors, while providing leeway for new compounds. Antitargets allow to profile the ligands according to five metabolizing and drug-effluxing enzymes and proteins.
García-Sosa A. T.,* Maran, U., "Improving the Use of Ranking in Virtual Screening against HIV-1 Integrase with Triangular Numbers and Including Ligand Profiling with Antitargets", Journal of Chemical Information and Modeling, 2014, Vol. 54, Iss. 11, 3172-3185. DOI: http://dx.doi.org/10.1021/ci500300u, [PDF].
Anti-targets, ligand efficiency, and docking for HIV-1 RT, including some possible metabolic effects
We used ligand binding efficiencies in virtual screening, together with anti-targets composing several proteins involved in compound metabolism, in order to discover candidate compounds for wild-type and drug-resistant HIV-1 reverse transcriptases that have the best profiles, both for binding to a variety of targets (target proteins in several conformational, mutational, and hydration states), as well as their predicted interaction profile with PXR, SULT, and several CYPs.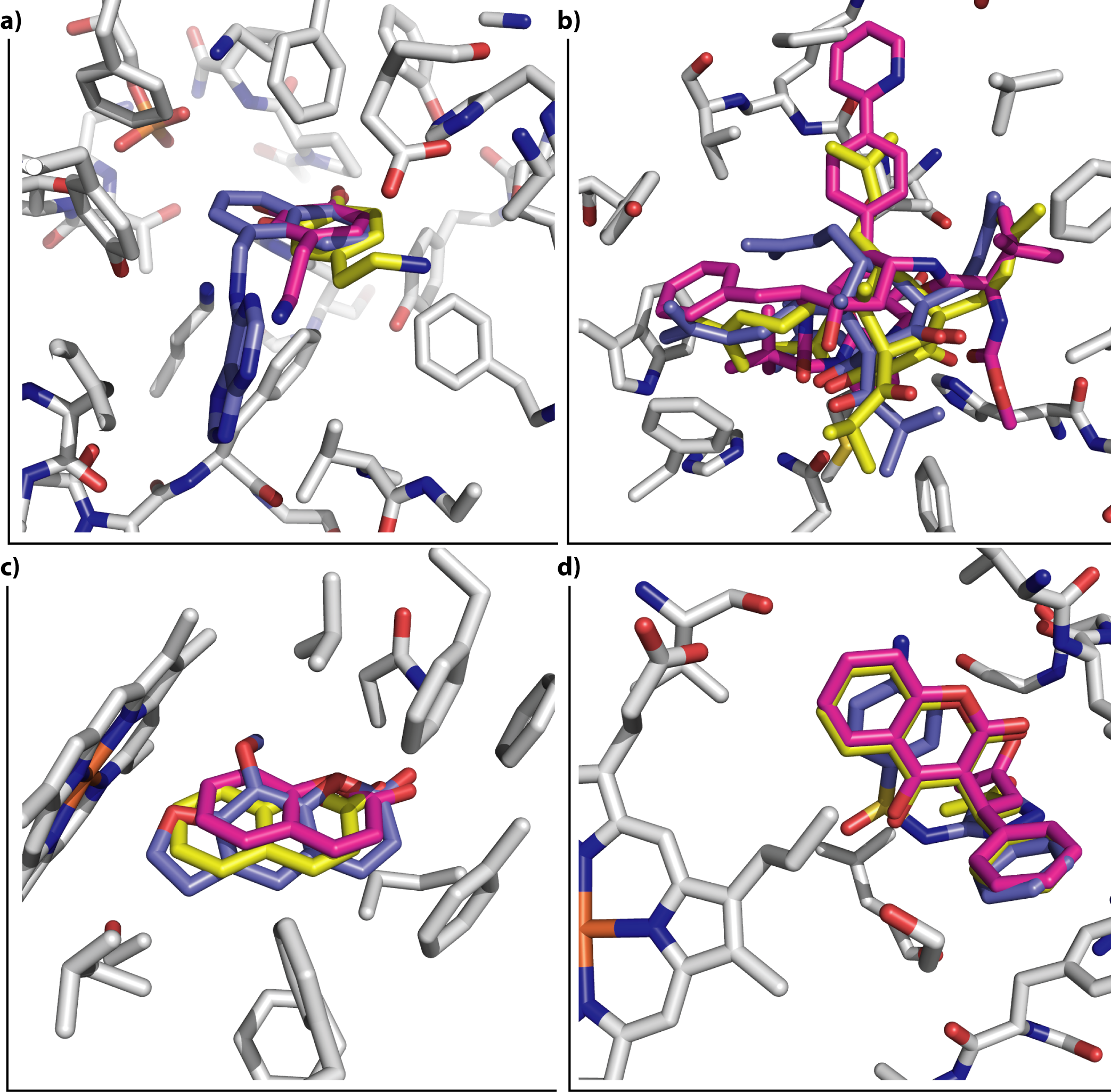
Predicted positive and negative interaction profiles, both for pharmaceutical target inhibition and for the metabolic in silico profile of candidate compounds are thus developed. This may help in polypharmacology, systems pharmacology, drug repurposing, and other settings involving optimization to a variety of partners and chemical properties.
García-Sosa A. T.,* Sild S., Takkis K., Maran U., "Combined Approach using Ligand Efficiency, Cross-Docking, and Anti-Target Hits for Wild-Type and Drug-Resistant Y181C HIV-1 Reverse Transcriptase", Journal of Chemical Information and Modeling, 2011, Vol. 51, Iss. 10, 2595-2611.
link: HTML, DOI, [PDF].
See Publications.
Hydration of ligands in 2,332 crystal structures and their associated binding energies
Recent findings are changing the view of water molecules in binding events, from a passive role to a fundamental and driving one. They can bridge protein-ligand interactions and guide their specificity, energetics, both enthalpically and entropically, and these outcomes depend on the shape, size, chemical, and dynamical nature of protein binding site and ligands. High resolution X-ray crystal structures with reported experimentally determined Ki or Kd show that there is no statistically significant difference in binding energy between binding sites with tightly-bound water molecules and those without them. Other physicochemical properties and ligand efficiencies are also compared. There are also benefits of lower logP and better developability for tightly hydrated compounds, while stronger potency is not always required or beneficial. These results also hold for drugs/non-drugs comparisons.In addition, agonists and antagonists that use tightly bound water bridges are smaller, less lipophilic, and less planar; have deeper ligand efficiency indices; and in general, possess better physicochemical properties for further development. Therefore, tightly bound, bridging water molecules may in some cases be replaced and targeted as a strategy, though sometimes keeping them as bridges may be better from a pharmacodynamic perspective. The hydrated binding site may be one of the many structure conformations available to the receptor, and different ligands will have a different ability to select either tightly hydrated or non-tightly hydrated or non-hydrated receptor binding site conformations.
Compounds may thus be designed, and if a tightly bound, bridging water molecule is observed in the binding site, attempts to replace it should only be made if the subsequent ligand modification would improve also its ligand efficiency, enthalpy, specificity, and pharmacokinetic properties. If the modification does succeed in replacing the tightly bound, bridging water molecule, it will have at least achieved benefits for ligand optimization and development independently of either positive or negative change in binding affinity outcome.

García-Sosa A. T.,* "Hydration Properties of Ligands and Drugs in Protein Binding Sites: Tightly-Bound, Bridging Water Molecules and Their Effects and Consequences on Molecular Design Strategies" , Journal of Chemical Information and Modeling, 2013, Vol. 53, Iss. 6, 1388-1405. DOI: http://dx.doi.org/10.1021/ci3005786, [Abstract], [PDF].
See Publications.
DrugLogit: Multivariate Logistic Regression, Drugs, Non-drugs, and Disease-Specificity
Simple and chemically intuitive logistic functions have been built to distinguish between drugs and non-drugs, taking into account individual disease categories or organ groups. These relate a few readily-available physicochemical molecular properties to a continuous and gradual outcome being the probability of classification as a drug, or drug-likeness.
See Calculators Using These Equations.
García-Sosa A. T.,* Oja M., Hetényi C., Maran U., "DrugLogit: Logistic Discrimination between Drugs and Nondrugs Including Disease-Specificity by Assigning Probabilities Based on Molecular Properties", Journal of Chemical Information and Modeling, 2012, Vol. 52, Iss. 8, 2165-2180. DOI: http://dx.doi.org/10.1021/ci200587h, [Abstract], [PDF].
See Publications.
Description and characterization of drugs and non-drugs in chemical space through principal components analysis, and through probability density functions
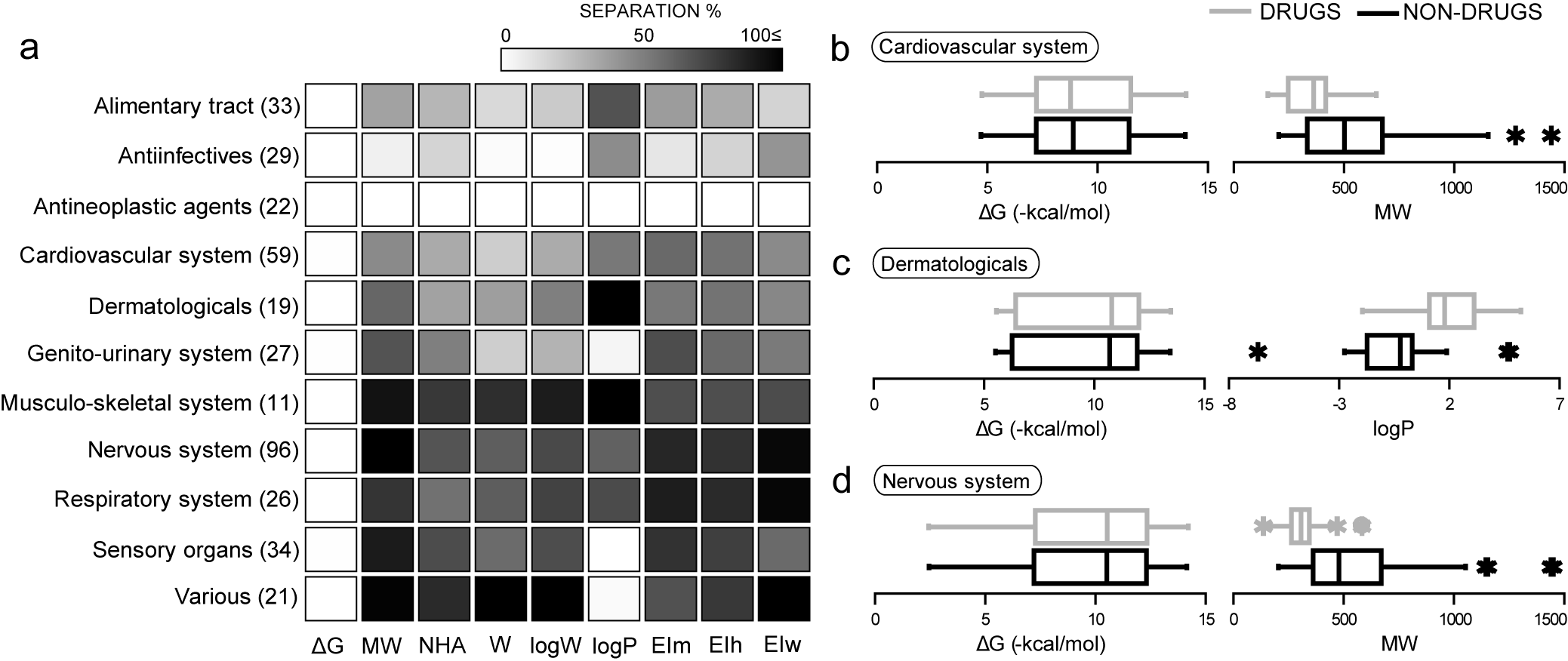
[PubMed 22376034], [DOI], [HTML], [PDF].
García-Sosa A. T.,* Oja M., Hetényi C., Maran U., "Disease-specific differentiation between drugs and non-drugs using principal component analysis of their molecular descriptor space", Molecular Informatics, 2012, Vol. 31, 369-383.
[DOI], [HTML], [PDF].
García-Sosa A. T.,* and Maran U., "Drugs, non-drugs, and disease category specificity: Organ effects by ligand pharmacology", SQER, 2013, Vol. 24, 585-597.
[DOI], [HTML], [PDF].
See Publications.
2011: Named 'Hot Article in Biochemistry' by Wiley
Chemically Relevant Functional Group Substitutions of a Tightly-Bound Water Molecule Including Enthalpic, Entropic, Fully Explicit Solvent and Ground State Effects
New, modified ligands for the Abl tyrosine kinase-SH3 domain implicated in chronic myelogenous leukemia (CML, a cancer of white blood cells) were built by generating different scenarios: including, neglecting, and targeting a specific hydration site with several chemical functional groups. This enabled determining the thermodynamic and structural effects of these chemical probes in a protein-ligand complex that has a tightly-bound water molecule bridging the interaction. Molecular dynamics using thermodynamic integration (a relative of the free energy perturbation method), explicit water, periodic boundary conditions, full thermodynamic cycles to include the desolvation effects of ligands as well as of the individual water molecule, and correcting for the ground state provides valuable information on the different groups that are best suited chemically and physically for substitution. Some water molecules are loosely bound to the protein or ligand surface, and so will not interfere or interact strongly. However, there are some which will be strongly bound and can be distinguished in several complexed states of the protein (see WaterScore below). This paper shows the different routes available for modification of the ligand structure based on a strongly-bound water, and their effects and insight into ligand optimization of protein–ligand–water systems.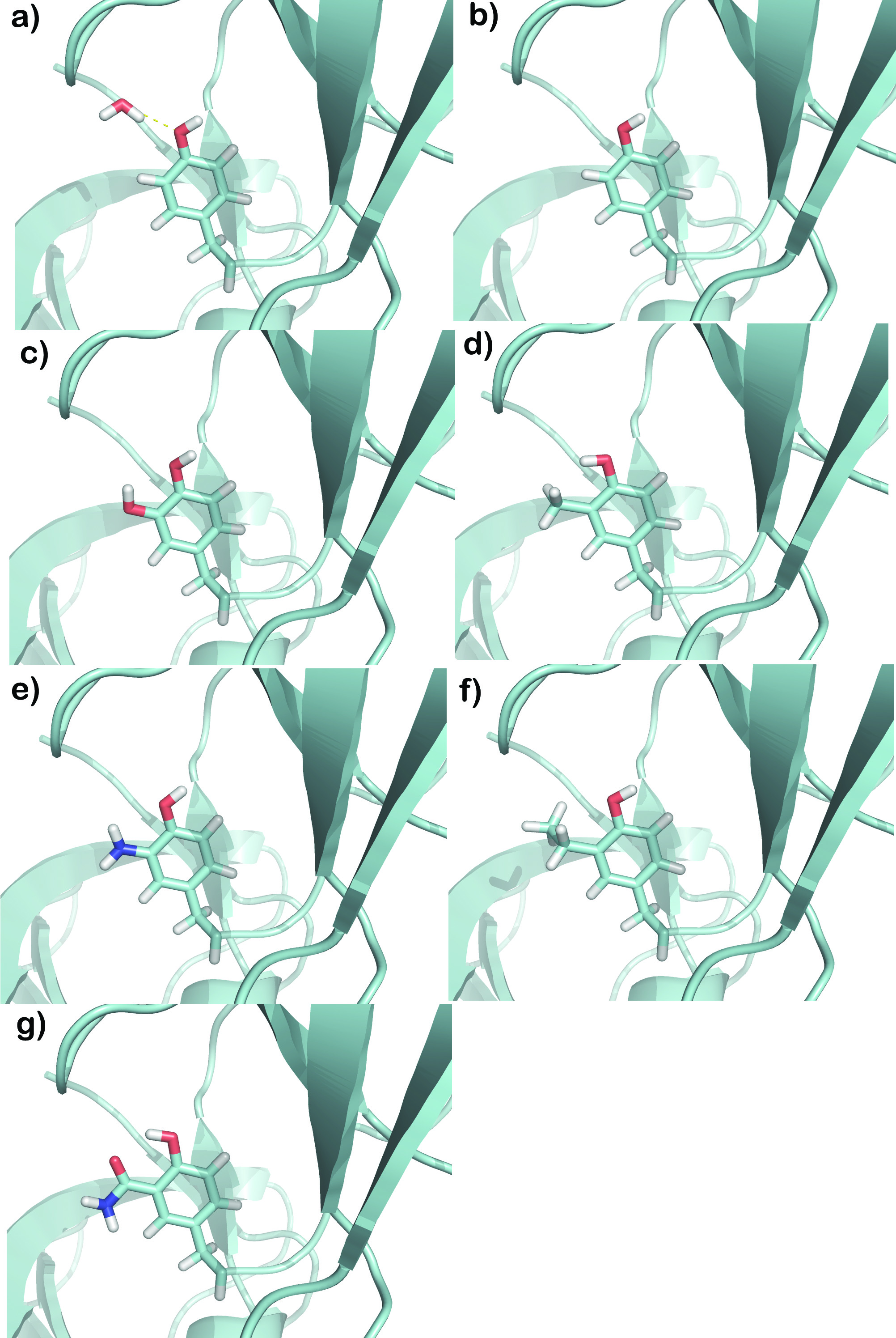
The energy of extracting a water molecule from the bulk solvent of water molecules has the reverse sign, but the same magnitude, as the energy of replacing a water molecule into the bulk solvent.
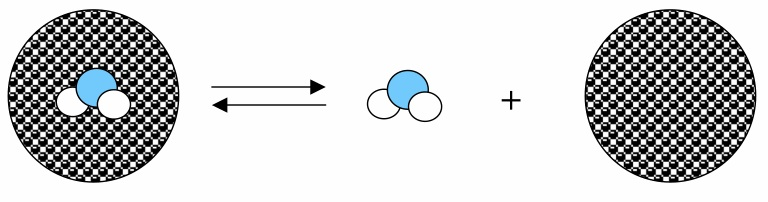
García-Sosa A. T.* and Mancera R.L., "Free Energy Calculations of Mutations Involving a Tightly Bound Water Molecule and Ligand Substitutions in a Ligand-Protein Complex", Molecular Informatics, 2010, Vol. 29, Iss. 8-9, 589-600.
link: HTML, DOI, [PDF].
(typo: p 594, l 14 should read "atomic fluctuation = 0.89 Å, effective k = 2 kcal / mol / Å ")
See Publications.
Ligand and drug efficiency indices (EI)
Efficiency indices effectively normalize the free energy of binding of a drug or ligand per a given measure of that compound's molecular characteristics, such as molecular weight or number of heavy atoms. Particular ligand efficiencies were determined to improve the correlation between experimental and calculated EI values for protein-drug complexes. ΔG/W (free energy of binding divided by Wiener index, a topological measure), ΔG/NoC (free energy of binding divided by number of carbons, a measure of size and lipophilicity), and ΔG/P (free energy of binding divided by the octanol / water partition coefficent, a measure of lipophilicity and hydrophilicity) produced improved correlations for EI for several docking programs and scoring functions. Better correlations between experimental and calculated EI values can improve the accuracy of virtual screening and molecular docking. In addition, the common bias of scoring functions in favor of larger ligands can be removed.
García-Sosa A. T.,* Hetényi C., Maran U., "Drug Efficiency Indices for Improvement of Molecular Docking Scoring Functions", Journal of Computational Chemistry, 2010, Vol. 31, Number 1, 174-184.
link: DOI , HTML, [PDF], [authorPDF].
García-Sosa A. T.,* Sild S., Maran U.,"Docking and Virtual Screening Using Distributed Grid Technology", QSAR & Combinatorial Science, 2009, Vol. 28, Number 8, 815-821.
link: DOI , HTML, [authorPDF].
See Publications.
Inhibitor design targeting H5N1 avian influenza
Drug design was conducted together with virtual screening for generating inhibitors that simultaneously occupy several binding sites of both wild-type H5N1 avian influenza neuraminidase, as well as of a drug-resistant mutant. Several ligand efficiency (also called binding efficiency) values were used to better characterize the compounds.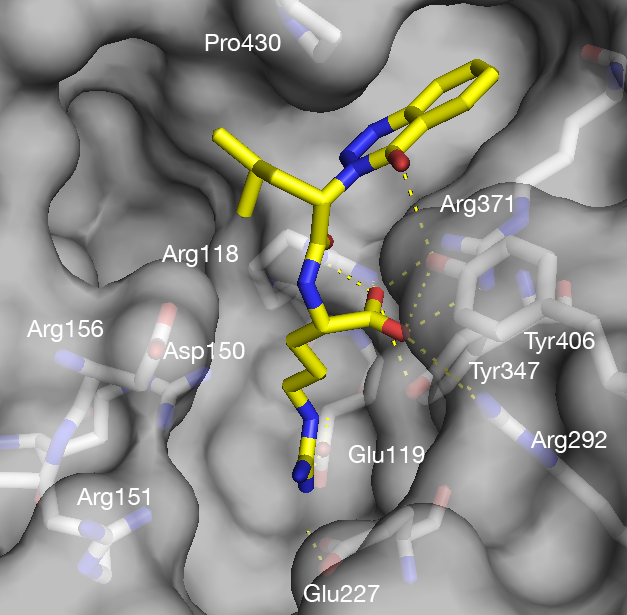
See Animation.
García-Sosa A. T.,* Sild S., Maran U., "Design of Multi-Binding-Site Inhibitors, Ligand Efficiency, and Consensus Screening of Avian Influenza H5N1 Wild-Type Neuraminidase and of the Oseltamivir-Resistant H274Y Variant", Journal of Chemical Information and Modeling, 2008, Vol. 48, 2074-2080.
link: DOI , Supporting Information , [PDF].
See Publications.
Drug design with metalloproteins, zinc centres and multiple molecular dynamics simulations
Small molecule inhibitors of Botulinum neurotoxin serotype A (BoNTA) were designed using multiple molecular dynamics simulations (MMDS) and a non-bonded four coordination active site zinc model, as well as projects on protein-ligand free energy of binding and other thermodynamic analyses of multiple protein-ligand systems.The use of 20 molecular dynamics simulations of at least 2 ns in explicit solvent results in improved sampling that provides a good measure to discriminate if a ligand in a given binding mode has favourable interactions inside a binding site, with the best ligands remaining in a strong coordination to the protein and having favourable interaction energy terms.
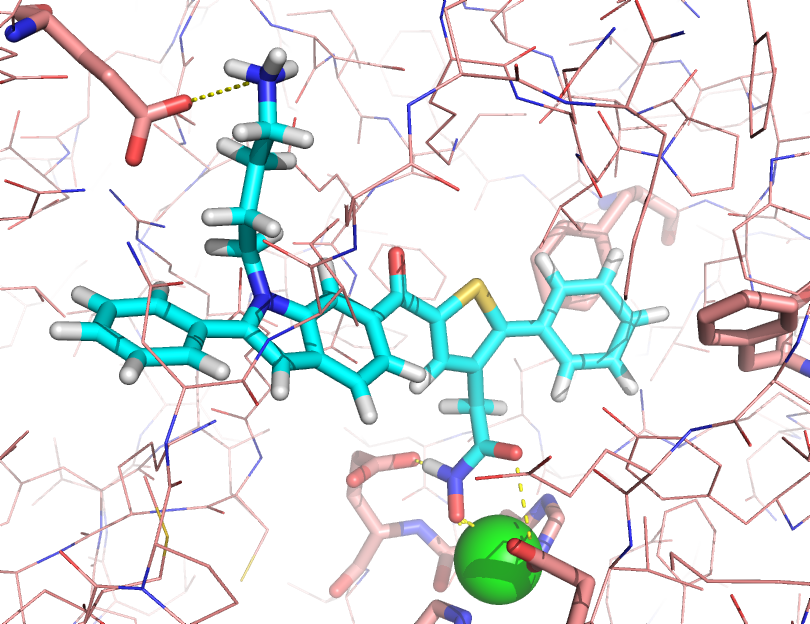
This page has a movie that shows part of an MD simulation of the 12 micromolar inhibitor of BoNTA. The zinc atom center is shown as a sphere in magenta:
See Movie.
Park J.G., Sill P.C., Makiyi E.F., Garcia-Sosa A. T., Millard C.B., Schmidt J.J., Pang Y.P., "Serotype-selective, Small-molecule Inhibitors of the Zinc Endopeptidase of Botulinum Neurotoxin Serotype A", Bioorganic & Medicinal Chemistry, 2006, Vol. 14, 395-408.
link: DOI , [PDF].
See Publications.
Explicit, crystallographic water molecules in computer-aided drug design
Hydration of protein-substrate/inhibitor complexes is a factor involved in the design of ligands. Water molecules have a very important role in the molecular recognition process as well as in the overall thermodynamics of protein-ligand binding and biomolecular processes. Therefore, dynamic simulation studies of proteins in an aqueous environment are critical for a realistic description of their structure and thermodynamic properties. A study of both the structural and dynamical properties of these systems proves useful to understand ligand–binding phenomena.A first step in these studies was a comprehensive survey of patterns in hydration sites as seen in the crystal structures of sets of proteins of interest. A multivariate function was developed to fit the properties of the water molecules observed both in the X ray crystal structures of the active sites of apo proteins and in the structure of the actives sites of complexes of the same proteins, to distinguish them from the properties of those water molecules not matched between sites. These comparisons were carried out for a variety of proteins. The predicted outcome variable (called WaterScore value) may be used to assign the probability of observing a particular water molecule from the apo protein structure in a structure of a complex of that protein, provided that it is not displaced sterically by the ligand or by a protein conformational change. The group of water molecules used in the analysis are those in the hydration shell of the ligand in the complex structure of the protein. This is interesting for drug design, docking strategies and methods because it allows the inclusion of a set of explicit water molecules, since the bound water molecules can be scored very easily and fast. The inclusion of specific water molecules allows for a better description of protein-ligand binding and improves the calculation of binding poses, intermolecular interactions and binding energy.
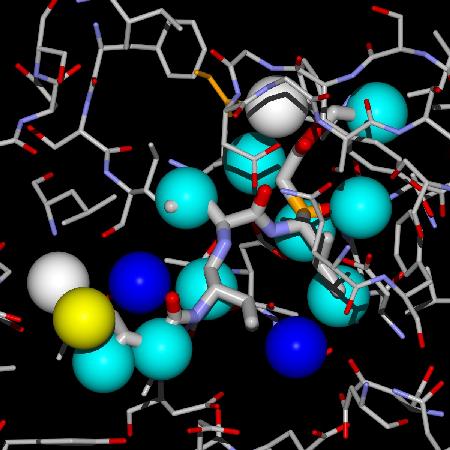
You can use the WaterScore Calculator© below to type in the values of the properties of water molecules and compute their WaterScore value. SCSA is the solvent accessible contact surface area (in Å2), and NPAC is the number of protein atomic contacts the water molecule has within a distance of 3.5 Å.
WaterScore Calculator ©
Note: A value of NaN indicates a very small value, close to zero. You need to have JavaScript enabled on your web browser. As a rough guide, a completely exposed water molecule would have a surface area of approx. 24.63 Å2.
García-Sosa A. T.,* Mancera R. L., Dean P. M. "WaterScore: A Novel Method for Distinguishing between Bound and Displaceable Water Molecules in the Crystal Structure of the Binding Site of Protein-Ligand Complexes", Journal of Molecular Modeling, 2003, Vol. 9, Issue 3, 172-182.
(copyright Springer-Verlag 2003). The original publication is available at this link: metapress , or by digital object identifier DOI, [PDF].
See Publications.
Tight waters in pharmacophores
Tightly-bound water molecules were also found to to be neccessary to explain pharmacophore binding model projection points. This showed that active ligands used tightly bound water molecules as interaction groups inside protein binding sites.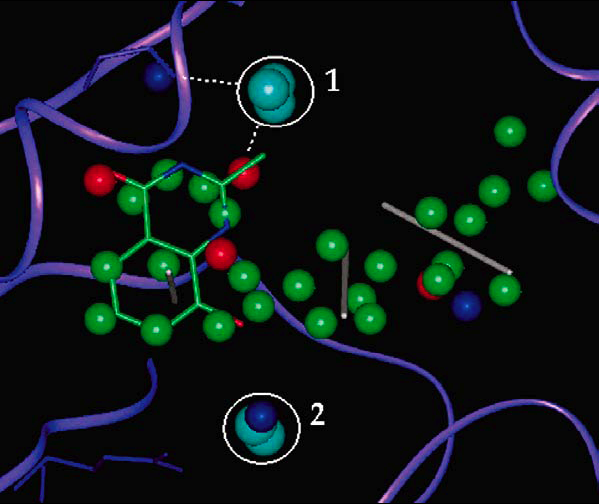
link: metapress, or DOI , [PDF].
See Publications.
PARP inhibitor design
Tightly bound water molecules have also been studied for their effects in ligand docking and de novo structure based drug design for several protein target cases.For example, poly(ADP-Ribose) polymerase, PARP:

García-Sosa A. T.,* Firth-Clark S., Mancera R. L. "Including Tightly-Bound Water Molecules in De Novo Drug Design. Exemplification Through the In Silico Generation of Poly (ADP-Ribose) Polymerase Ligands", Journal of Chemical Information and Modeling, 2005, Vol. 45, 624-633.
DOI , [HTML], [PDF].
See Publications.
CDK2 inhibitor design
In the case of Cyclin dependent kinase, CDK2: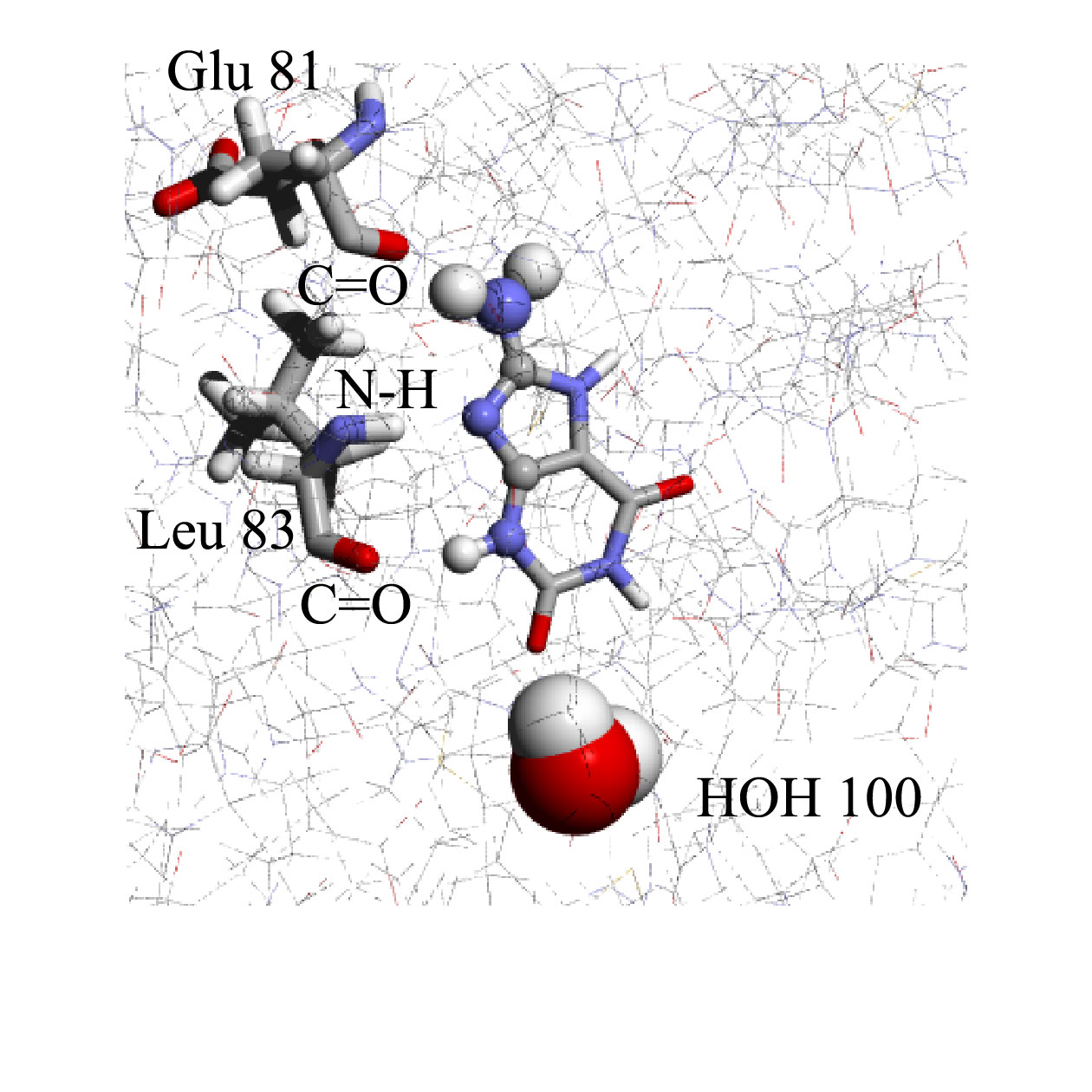
García-Sosa A. T.* and Mancera R. L. "The Effect of a Tightly-Bound Water Molecule on Scaffold Diversity in the Computer-Aided de novo Ligand Design of CDK2 Inhibitors", Journal of Molecular Modeling, 2006, Vol. 12, Issue 4, 422-431.
The original publication is available at this link: metapress , or by digital object identifier: DOI , [PDF].
See Publications.
Ab initio studies of Iron-Oxygen systems, FenOn+,-
During my undergraduate studies, I did research on transition metal-ligand systems with ab initio methods such as DFT.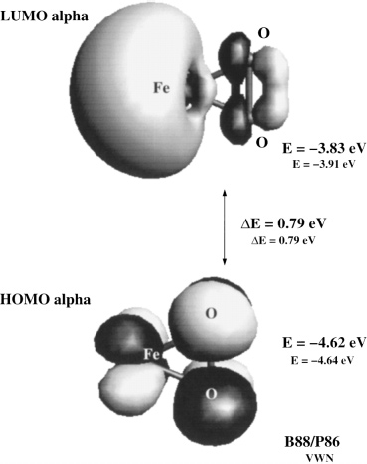
![A.T. Garcia-Sosa, M. Castro, A Density Functional Study of FeO2, FeO2+, and FeO2-, International Journal of Quantum Chemistry, 2000, Vol. 80, Issue 3, 307-319 Iron oxygen DFT vibration frecuency frequencies Abstract The lowest energy structures of FeO<sub>2</sub> were determined by means of density functional theory techniques as implemented in the program DGauss 3.0.1.
The calculations performed were of the all-electron type using two levels of theory, namely the local spin density approximation with the use of the Vosko–Wilk–Nusair (VWN) functional and the generalized gradient approximation (GGA) in the form of the Becke (1988) exchange and Perdew (1986) correlation functionals.
Results were visualized with UniChem.
Bond distances and angles as well as total energies were calculated for several states of the moieties: Fe(O)<sub>2</sub>, <em>C</em><sub>2<em>v</em></sub>; Fe(O)<sub>2</sub>, <em>D</em><sub>∞<em>h</em></sub>; Fe(η<sup>2</sup>-O<sub>2</sub>), <em>C</em><sub>2<em>v</em></sub>; Fe(η<sup>1</sup>-O<sub>2</sub>), <em>C</em><sub><em>s</em></sub>; and Fe(η<sup>1</sup>-O<sub>2</sub>), <em>C</em><sub>∞<em>v</em></sub>.
Molecular orbital and harmonic vibrational analyses were carried out for these species, in addition to Mulliken population analyses.
Singly positive and negative charged species were also considered and fully geometry optimized in a self-consistent field (SCF) gradient method.
Accurate ionization potentials and electron affinities (both vertical, <em>v</em>, and adiabatic, <em>a</em>, determinations) were thus able to be computed.
The results show the following for the ground state (GS) Fe(O)<sub>2</sub>, <em>C</em><sub>2<em>v</em></sub>, <em>M</em>=3: ∠OFeO=138.1° (133.6°) [values in parentheses are for local spin density approximation (LSDA)–VWN, while the others are at the GGA-B88/P86 level].
Here, <em>R</em><sub><em>e</em></sub> Fe–O=1.60 Å (1.57 Å), ET=−1414.2064 au (−1,410.5047 au), EA<sub><em>a</em></sub>=2.47 (2.60) eV, IP<sub><em>a</em></sub>=10.6 (10.5) eV, EA<sub><em>v</em></sub>=2.41 (2.20) eV, IP<sub><em>v</em></sub>=10.67 (10.63) eV, and EA<sub>exp</sub>=2.349 eV (in agreement with related studies).
In the GS the dioxygen molecule is found to be dissociated, compared to those states which have coordination modes where the O<sub>2</sub> molecule formally persists.
A 3<em>d</em>4<em>sp</em> configuration for the iron atom is found to be especially relevant in Fe–O bond formation.
The iron–oxygen and oxygen–oxygen bonds involved are characterized.
A direct relationship is observed between these electronic and structural properties, influencing also the total energy for a given molecule.](webfigFeO2bb.jpg)
García-Sosa A. T. and Castro M., "A Density Functional Study of FeO2, FeO2+, and FeO2- ", International Journal of Quantum Chemistry, 2000, Vol. 80, Issue 3, 307-319.
link: HTML , or DOI , [PDF] , [TEX].